Journal of Molecular Biology
ABSTRACT
Glioma is a type of tumor originating from glial cells in the brain and spinal cord. It primarily affects astrocytes, oligodendrocytes, and ependymal cells. Mutations in the Isocitrate Dehydrogenase 1 (IDH1) and Isocitrate Dehydrogenase 2 (IDH2) genes are pivotal in gliomagenesis, particularly in diffuse lower-grade gliomas and secondary glioblastomas. These mutations, especially at R132 in IDH1 and R172 in IDH2, confer a neomorphic activity converting α-ketoglutarate (α-KG) to D-2-hydroxyglutarate (2-HG). Elevated 2-HG disrupts α-KG–dependent dioxygenases, resulting in genome-wide epigenetic modifications, collectively termed the glioma CpG island methylator phenotype (G-CIMP). Clinically, these mutations correlate with younger age at diagnosis, favorable prognosis, and improved survival. IDH mutations are now essential in the 2021 WHO classification of CNS tumors. Targeted therapies such as ivosidenib, enasidenib, and vorasidenib aim to reverse 2-HG accumulation. This commentary outlines the molecular basis, clinical impact, and therapeutic opportunities related to IDH1/2 mutations in glioma.
Keywords: IDH1, IDH2, glioma, 2-hydroxyglutarate, epigenetic dysregulation, G-CIMP, targeted therapy.
INTRODUCTION
Metabolic gene mutations are increasingly recognized as key drivers of tumorigenesis across various cancer types, including gliomas. These mutations impair normal cellular metabolism and disrupt crucial regulatory processes that maintain epigenetic stability. In particular, genes such as Fumarate Hydratase (FH) and Succinate Dehydrogenase Complex Subunit C (SDHC) have been implicated in a subset of hereditary and sporadic cancers, where their loss leads to the accumulation of metabolic intermediates with oncogenic potential [1 - 2].
However, among the metabolic genes implicated in glioma, isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) stand out due to the frequency and specificity of their mutations in lower-grade gliomas and secondary glioblastomas. These mutations are most commonly missense mutations at arginine residue 132 in IDH1 and residue 172 in IDH2, resulting in a neomorphic enzymatic function. Instead of catalyzing the conversion of isocitrate to α-ketoglutarate (α-KG), the mutant enzymes reduce α-KG to an oncometabolite, D-2-hydroxyglutarate (2-HG) [3 - 4].
The accumulation of 2-HG interferes with a broad class of α-KG–dependent dioxygenases, including histone and DNA demethylases, leading to widespread epigenetic alterations. This epigenetic dysregulation contributes to abnormal cellular differentiation and promotes a pro-tumorigenic environment [5 - 6]. As a result, IDH mutations are now considered not only a hallmark of glioma pathogenesis but also a defining molecular feature that helps classify gliomas into distinct biological subtypes. The discovery of these mutations has profoundly reshaped our understanding of glioma biology and opened new avenues for targeted therapy.
Pathophysiology
Mutations in IDH1 and IDH2 fundamentally rewire cellular metabolism and epigenetic regulation, profoundly altering the biological landscape of glioma. One of the most critical downstream effects of these mutations is the excessive production of the oncometabolite D-2-hydroxyglutarate (2-HG), which mimics the structure of α-ketoglutarate (α-KG) and competitively inhibits a wide range of α-KG–dependent dioxygenases [5].
These enzymes play pivotal roles in regulating the epigenome, particularly histone demethylases and Ten-Eleven Translocation (TET) DNA demethylases, which are responsible for removing methyl groups from DNA and histone proteins. Inhibition of these dioxygenases by 2-HG results in widespread hypermethylation of DNA and histones, leading to transcriptional silencing of genes involved in cellular differentiation and tumor suppression. This hypermethylation pattern is a defining feature of the glioma CpG Island Methylator Phenotype (G-CIMP), which is strongly associated with IDH-mutant gliomas [7 - 8].
Experimental studies have further illuminated the role of mutant IDH in altering normal brain development. In mouse models and neural stem cells engineered to express mutant IDH1, researchers have observed disrupted neural differentiation, abnormal maintenance of stem-like states, and significant alterations in chromatin architecture [9]. These changes not only block maturation pathways but also create a permissive environment for tumor initiation and progression.
Another key consequence of 2-HG accumulation is the induction of a pseudohypoxic state. Under normal conditions, the α-KG–dependent prolyl hydroxylases regulate the stability of hypoxia-inducible factor 1-alpha (HIF-1α) by marking it for degradation. However, 2-HG inhibits these hydroxylases, resulting in stabilization of HIF-1α, even in normoxic conditions [9]. This stabilization enhances the transcription of hypoxia-responsive genes involved in angiogenesis, such as Vascular Endothelial Growth Factor (VEGF), thereby promoting the formation of new blood vessels to support tumor growth and survival.
Collectively, these interconnected molecular alterations underscore how IDH mutations act as early driver events in gliomagenesis, reshaping both the metabolic and epigenetic landscape to favor tumor development and resistance to differentiation.
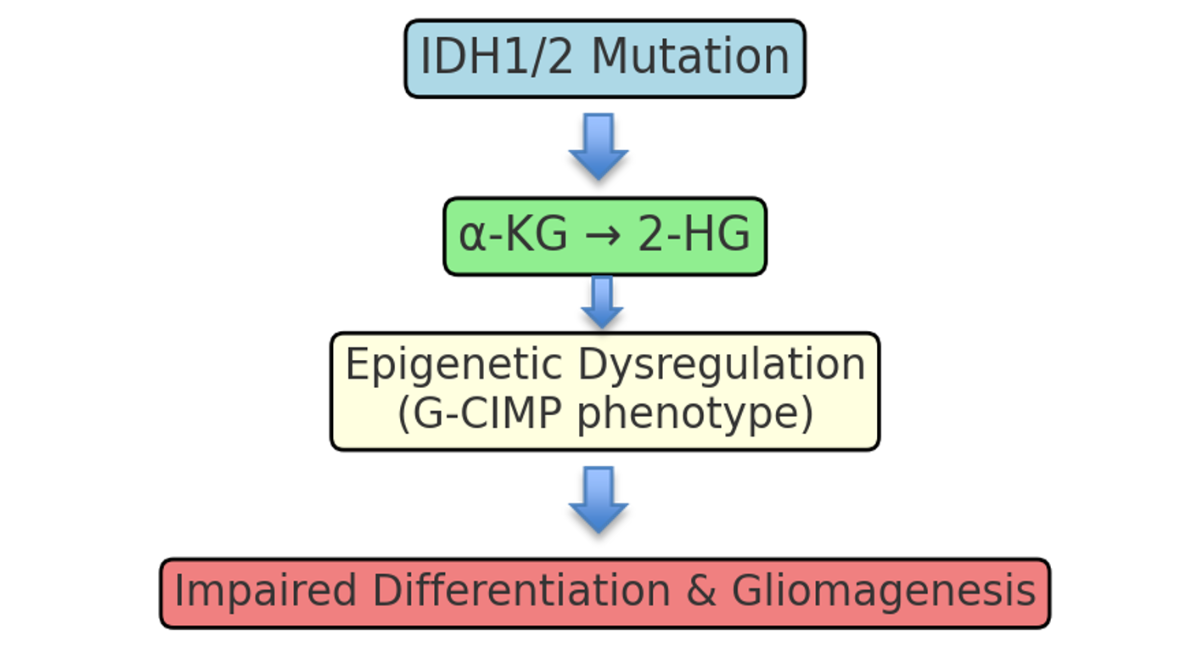
Figure 1: Pathogenic mechanism of IDH1/2 mutations in glioma.
Clinical Significance
Mutations in IDH1 and IDH2 are not only biologically significant but also carry substantial clinical implications in the diagnosis, classification, and management of gliomas. These mutations are highly prevalent in lower-grade gliomas (WHO grade II and III) and secondary glioblastomas, occurring in approximately 70–80% of such cases. In contrast, they are rare in primary glioblastomas, which tend to follow a different molecular and clinical course [10].
One of the most striking features of IDH-mutant gliomas is their association with younger patient age at diagnosis, typically presenting in the third or fourth decade of life. Additionally, patients with IDH-mutant gliomas generally have a more favorable prognosis, with significantly longer progression-free and overall survival compared to their IDH-wildtype counterparts [11]. This prognostic benefit is attributed to the slower growth kinetics of IDH-mutant tumors, as well as their unique molecular characteristics, including G-CIMP hypermethylation and reduced cellular proliferation.
The 2021 World Health Organization (WHO) Classification of Central Nervous System Tumors has recognized IDH mutation status as a central diagnostic criterion, effectively redefining how gliomas are classified. For instance, the diagnosis of astrocytoma or oligodendroglioma now explicitly requires the presence of an IDH mutation, distinguishing them from IDH-wildtype glioblastomas, which follow a more aggressive course and require different therapeutic strategies [10].
From a clinical management perspective, IDH mutation testing has become an essential tool for guiding decision-making. It assists not only in diagnosis and prognostication but also in monitoring disease progression, as IDH-mutant gliomas may follow a more indolent yet prolonged clinical trajectory. Moreover, the presence of an IDH mutation may influence the choice of therapy, particularly as mutant-specific IDH inhibitors (e.g., vorasidenib, ivosidenib) move toward broader clinical use [12].
As precision medicine continues to evolve, the IDH1/2 mutation status exemplifies how a single molecular alteration can influence every stage of clinical care—from diagnosis and risk stratification to treatment selection and long-term follow-up.
Therapeutic Implications
The discovery of IDH1 and IDH2 mutations has not only transformed the molecular classification of gliomas but also opened the door to targeted therapeutic strategies. One of the most promising approaches involves the use of mutant-specific IDH inhibitors, which aim to counteract the harmful effects of the oncometabolite 2-hydroxyglutarate (2-HG).
Agents such as ivosidenib (AG-120), which targets mutant IDH1, and enasidenib (AG-221), which targets mutant IDH2, were initially developed and approved for acute myeloid leukemia (AML), where IDH mutations also play a pathogenic role [13]. Their success in hematologic malignancies has led to their investigation in solid tumors, including gliomas, where the production of 2-HG leads to epigenetic dysregulation and tumor progression.
More recently, vorasidenib, a dual inhibitor of mutant IDH1 and IDH2 that is brain-penetrant, has shown particularly encouraging results in patients with IDH-mutant lower-grade gliomas. In a landmark phase III clinical trial, vorasidenib demonstrated significantly prolonged progression-free survival compared to placebo in patients with residual or recurrent grade II gliomas who had undergone surgery but had not yet received radiotherapy or chemotherapy [12]. Importantly, the drug was well tolerated, and many patients were able to delay more aggressive treatments such as radiation, which can have long-term neurocognitive side effects.
The rationale behind IDH inhibition is to reduce intracellular 2-HG levels, thereby allowing restoration of normal DNA and histone demethylation activity, reactivation of tumor suppressor genes, and reinitiation of normal differentiation pathways. By reversing the epigenetic blockade caused by mutant IDH enzymes, these therapies aim not only to slow tumor growth but potentially to shift tumor biology toward a less aggressive phenotype.
Although these agents are still undergoing evaluation in large-scale clinical trials, they represent a significant leap toward precision oncology in glioma care. Future studies will likely refine their use in combination with chemotherapy, radiotherapy, or immunotherapy, potentially enhancing efficacy and extending survival in this patient population.
CONCLUSION
Mutations in IDH1 and IDH2 play a central role in gliomagenesis by altering metabolism and driving epigenetic dysregulation through the production of 2-hydroxyglutarate (2-HG). These changes impair cellular differentiation and promote tumor development. Clinically, IDH mutations define a distinct glioma subtype associated with younger age, better prognosis, and are now a key component of the 2021 WHO CNS tumor classification.
Importantly, mutant-specific IDH inhibitors such as ivosidenib, enasidenib, and vorasidenib offer targeted treatment options, showing promise in slowing disease progression and delaying the need for aggressive therapies. As such, IDH mutations serve as both biomarkers and therapeutic targets, exemplifying the impact of precision medicine in neuro-oncology.
REFERENCES
- Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. The Journal of clinical investigation. 2013;123(9):3652-8. [ CrossRef] [ Google Scholor] [ PubMed]
- Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith Jr WI, Jahromi MS, Xekouki P, Szarek E. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer discovery. 2013;3(6):648-57. [ CrossRef] [ Google Scholor] [ PubMed]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H. IDH1 and IDH2 mutations in gliomas. New England journal of medicine. 2009;360(8):765-73. [ CrossRef] [ Google Scholor] [ PubMed]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739-44. [ CrossRef] [ Google Scholor] [ PubMed]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19(1):17-30.. [CrossRef] [Google Scholor] [PubMed]
- Turcan S, Rohle D, Goenka A, et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature, 483(7390), 479–483. [ CrossRef] [ Google Scholor] [ PubMed]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17(5):510-22. [ Google Scholor] [ PubMed]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012 Mar 22;483(7390):474-8. Nature. 2012;483(7390):474-8. [ CrossRef] [ Google Scholor] [ PubMed]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation.Nature. 2012;483(7390):484-8.Nature, 483(7390), 484–488. [ CrossRef] [ Google Scholor] [ PubMed]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-oncology. 2021 Aug 1;23(8):1231-51.. 2021;23(8):1231-51. Oncol. 2021;23(8):1231–1251. [ CrossRef] [ Google Scholor] [PubMed]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A. An integrated genomic analysis of human glioblastoma multiforme. science. 2008;321(5897):1807-12, 321(5897), 1807–1812. [ CrossRef] [ Google Scholor] [ PubMed]
- Mellinghoff IK, Van Den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, Mendez J, Yust-Katz S, Welsh L, Mason WP, Ducray F. Vorasidenib in IDH1-or IDH2-mutant low-grade glioma. New England Journal of Medicine. 2023;389(7):589-601. J Med. 2023;389(1):20–31. [ CrossRef] [ Google Scholor] [ PubMed]
- DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–2398. [ CrossRef] [ Google Scholor] [PubMed]
REFERENCES
Indexed In
DOAJ
CrossRef
PubMed
MEDLINE
ResearchBib
OAJI
Sindexs
EBSCO A-Z / Host
OCLC - WorldCat
Journal Flyer
