ABSTRACT
Background
Congenitally corrected transposition of the great arteries (CCTGA) is a rare congenital cardiac anomaly. Adult survival is uncommon due to progressive systemic right ventricular dysfunction, conduction abnormalities, and associated structural defects. Among these, aortic stenosis is an exceedingly rare association and is the focus of this case report.
Case Presentation
We report a 45-year-old woman with previously undiagnosed CCTGA who presented with syncope due to complete heart block. Evaluation revealed severe valvular aortic stenosis and a moderate ventricular septal defect with elevated pulmonary vascular resistance. Given the high surgical risk associated with systemic right ventricular dysfunction, a tailored transcatheter approach was planned.
Outcome
The patient underwent dual-chamber permanent pacemaker implantation followed by balloon aortic valvuloplasty. The procedure was successfully completed without complications, resulting in symptomatic improvement and stabilization. Surgical intervention was deferred due to prohibitive risk.
Conclusion
This case highlights the rare coexistence of severe aortic stenosis and complete heart block in adult CCTGA and emphasizes the importance of individualized, multidisciplinary decision-making. In high-risk patients, a combined transcatheter strategy may offer a safe and effective palliative alternative to surgery.
Keywords: Adult Congenital Heart Disease; CCTGA; Aortic Stenosis; Dextrocardia; Heart Block
INTRODUCTION
Congenitally corrected transposition of the great arteries (CC-TGA) is a rare congenital heart disease (CHD) characterized by atrioventricular and ventriculoarterial discordance, accounting for 0.4% of all CHD [1,2]. Although the circulation is physiologically corrected, the morphological right ventricle (RV) functions as the systemic ventricle, predisposing patients to progressive systemic ventricular dysfunction, tricuspid regurgitation (TR), and conduction abnormalities [3,4]. Associated lesions such as ventricular septal defect (VSD) and pulmonary outflow obstruction are common; however, valvular aortic stenosis (AS) is rarely reported [5-7]. We present a complex adult case of CCTGA with severe valvular AS and complete heart block managed with a tailored interventional approach.
CASE PRESENTATION
History
A 45-year-old female presented with a 4-month history of progressive dyspnea on exertion and a 1-month history of syncopal episodes. The dyspnea increase from NYHA class II to class III in the last one month. There was also history of paroxysmal nocturnal dyspnea. There was no significant past medical history. Her obstetric history included three pregnancies, with one vaginal delivery and two lower segment caesarean sections. All pregnancies were reportedly uneventful with no peripartum cardiac complications.
Examination
On examination, the patient was dyspneic, had a heart rate of 45 beats per minute, which was low volume and irregular. Blood pressure was 110/70 mmHg in the right upper limb, and oxygen saturation was 97% on room air.
Cardiovascular examination revealed raised JVP. The apex beat was in the right 5th intercostal space along the midclavicular line with a heaving character. Auscultation demonstrated a single S2, an ejection systolic murmur (grade 3/6) best heard in the right 2nd intercostal space, and a pansystolic murmur (grade 3/6) best heard in the right 3rd–4th parasternal area.
Investigations
Electrocardiogram and Chest X-ray
ECG demonstrated complete heart block. Chest X-ray revealed dextrocardia with cardiomegaly (Figure 1).
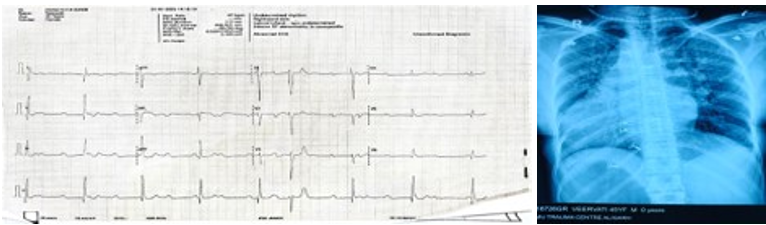
Figure 1: ECG and Chest X Ray of the Patient
Note: ECG shown reveals complete heat block, negative p wave in lead I and positive in II, III and aVF (situs inversus), poor R wave progression (dextrocardia). Chest x ray revealed situs inversus, dextrocardia and cardiomegaly.
Laboratory Findings
Blood investigations showed significantly elevated NT pro BNP levels (18,500 pg/mL), raised lactate levels (3.8 mmol/L) and low hemoglobin (9.2 g/dl).
Transthoracic Echocardiography
Echocardiography demonstrated situs inversus with dextrocardia and D-loop ventricles with atrioventricular–ventriculoarterial (AV–VA) discordance. The systemic ventricle (morphologic right ventricle) was dilated with reduced systolic function (TAPSE = 12 mm, RV fractional area change = 27%), along with severe tricuspid regurgitation. A perimembranous ventricular septal defect was also noted, partially restricted by the septal leaflet.
Additionally, there was severe right ventricular outflow tract obstruction in the form of aortic valve stenosis with a mean gradient of 48 mmHg. Cardiac computed tomography was done to confirmed the diagnosis as shown in figure 3.
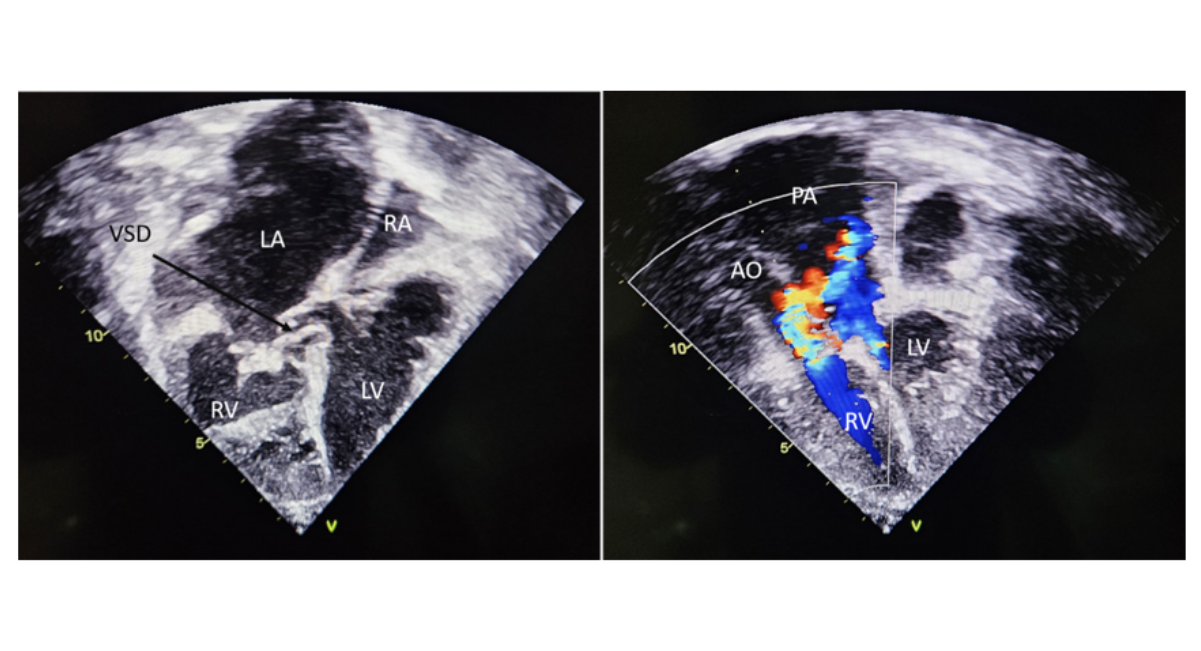
Figure 2: Transthoracic Echo Image of the Patient
Note: 2D echo image shows smooth walled left ventricle on the left and trabeculated right ventricle on the right (d looped in CCTGA due to situs inversus). There is a peri membranous VSD as shown by the black arrow. The thickened tricuspid valve can also be appreciated. The colour doppler echo image shows the great vessels, aorta and pulmonary artery. Flow acceleration can be appreciated across the aortic valve.
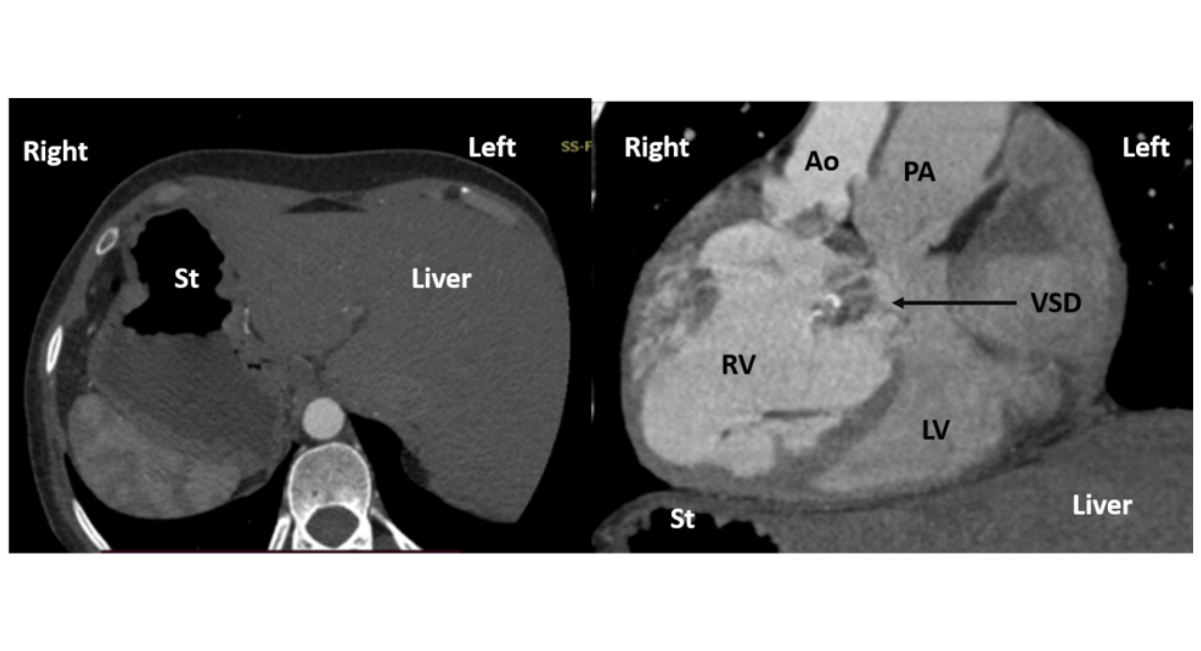
Figure 3: CT Scan Image of the Patient
Note: CT scan image on the left shows axial cut, showing stomach bubble on the right and liver on the left suggestive of abdominal situs inversus. CT scan image on the right shows sagittal cut, showing d loop ventricles, hypertrophied right ventricle and aorta to the right of pulmonary artery (d malposed aorta in CCTGA due to situs inversus).
The case was discussed with the surgical team and a combined decision was taken to do an oximetry analysis for the VSD, a balloon aortic valvotomy (BAV) and implant a pacemaker (PPI). Cardiac catheterization study revealed significant step up with Qp/Qs of 1.7 and PVR 5.1 wood unitsxm2. A dual chamber PPI was implanted, with the screwing lead placed in morphological LV and tined lead in morphologic RA. BAV was performed with Tyshak II balloon 16mm x4.0cm for annulus of 1.8cm and peak to peak gradient across the Aortic valve decreased from 69mm Hg to 14 mm Hg.
Following the procedure there was significant symptomatic relief, and RV systolic function showed improvement, (TAPSE = 20, RV-FAC = 40%). The patient was discharged in satisfactory condition on oral diuretic therapy. On, three and six months follow up, the patient showed good symptomatic relief, with no further episodes of syncope or dyspnea. Figure 4 shows the patient management timeline.
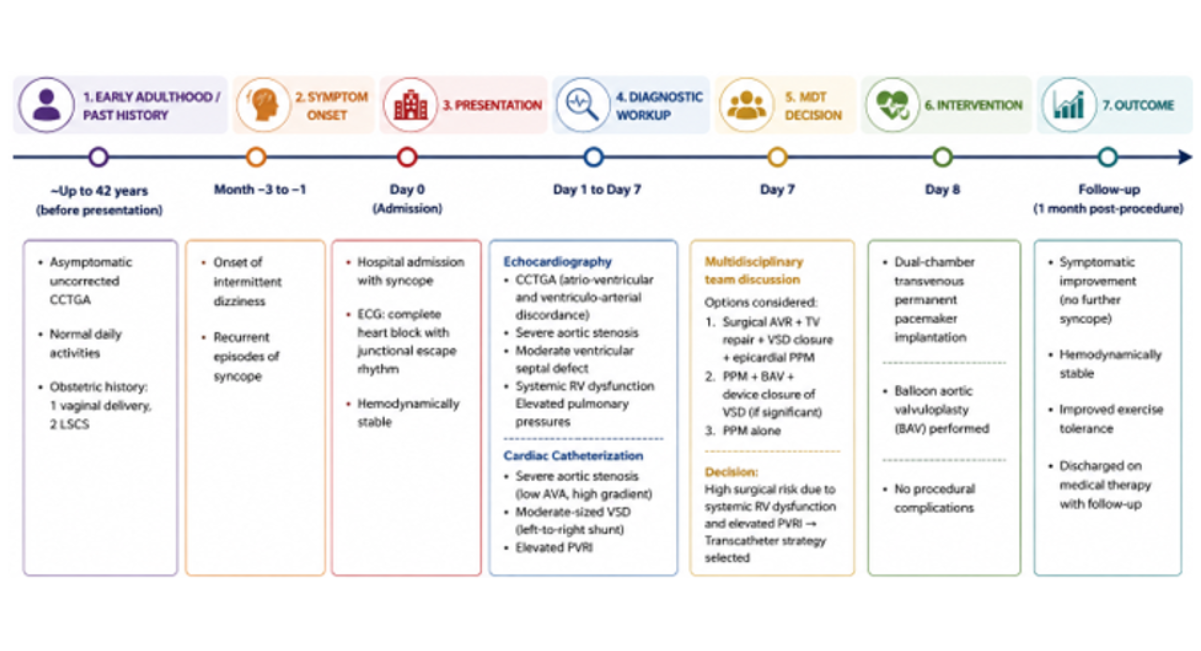
Figure 4: Figure Showing Patient Timeline
Note: Patient Timeline from Symptom Onset to Diagnosis, Intervention and Outcome
DISCUSSION
Survival into adulthood in patients with uncorrected CCTGA and associated cardiac defects is uncommon. This is largely due to early detection and timely intervention in developed countries, as well as attrition from complications such as systemic right ventricular failure and complete heart block. However, delayed diagnosis is still encountered in developing nations.
Our patient was a 45-year-old woman with previously undiagnosed CCTGA who had successfully completed three pregnancies, including one normal vaginal delivery and two cesarean sections. The late diagnosis of such a complex congenital heart disease highlights the challenges of early detection and the possibility of missed congenital cardiac lesions at the primary healthcare level.
Another unique aspect of our case is the rare combination of defects in an adult patient with CCTGA and the individualized treatment strategy adopted. Our patient presented with multiple coexisting lesions, including severe AS, a rare association in CCTGA, with only a handful of cases reported in the literature [6,7]. Syncope secondary to complete heart block made PPI mandatory. The principal management dilemma was whether to address the associated intracardiac defects, namely the VSD and AS.
An urgent multidisciplinary team (MDT) discussion was conducted, and three treatment options were considered:
(1) Surgical aortic valve replacement, tricuspid valve repair, VSD closure, and epicardial PPI;
(2) Transvenous PPI with BAV and
(3) Transvenous PPI alone.
The MDT decided against surgical intervention because of the high operative risk associated with systemic RV dysfunction and elevated PVRI. Cardiac catheterization was therefore planned to assess the severity of AS, quantify the VSD shunt, and proceed with pacemaker implantation. Hemodynamic assessment confirmed severe AS despite systemic RV dysfunction, along with a moderate-sized VSD and elevated PVRI. Consequently, the patient underwent BAV and dual-chamber permanent pacemaker implantation.
Although BAV in adults is generally considered a palliative procedure with less favorable long-term outcomes than valve replacement, it represented a reasonable strategy in this patient. Given the reduced life expectancy associated with CCTGA and systemic RV dysfunction (8,9), BAV provided symptomatic relief while avoiding the substantial risks of surgery.
As far as closure of the VSD was concerned, although the Qp:Qs ratio was >1.5, we decided against surgical closure. The primary reasons were the elevated PVRI, which significantly increased the operative risk, and the broader concern regarding the patient’s overall life expectancy in the setting of CCTGA in an adult. In such a scenario, the long-term benefit of VSD closure would likely be limited, and the potential risks of surgery outweighed the expected gains. Therefore, a conservative strategy was deemed more appropriate [8,9].
This case illustrates a rare and complex presentation of CCTGA with severe aortic stenosis and complete heart block in an adult. Given the high surgical risk, a palliative approach with balloon aortic valvotomy and dual-chamber PPI provided effective symptomatic relief and improvement in systemic RV function. Individualized, multidisciplinary management remains essential in such challenging cases.
CONCLUSION
Adult survival with uncorrected CCTGA associated with severe valvular AS is exceedingly rare. This case highlights the diagnostic and therapeutic challenges posed by such complex anatomy and emphasizes the importance of multidisciplinary decision-making. In carefully selected high-risk patients, a tailored transcatheter approach may provide meaningful symptomatic and hemodynamic benefit while avoiding the considerable risks of surgical intervention.
PATIENT CONSENT STATEMENT
The authors confirm that written informed consent was obtained from the patient for publication of this case report and any accompanying images. The patient has been informed that no personal identifiers will be disclosed, and every effort has been made to ensure confidentiality.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest regarding the publication of this case report. No financial or personal relationships influenced the work reported in this manuscript.
LEARNING OBJECTIVE
This case highlights a rare adult presentation of uncorrected CCTGA with an unusual combination of severe valvular AS and complete heart block. It adds to the limited literature on coexisting left-sided obstructive lesions in CCTGA and illustrates the challenges of managing multiple high-risk lesions in a single patient. Importantly, it demonstrates the role of individualized, multidisciplinary decision-making and supports a tailored transcatheter strategy—BAV combined with PPI—as a feasible alternative in selected high-risk surgical candidates.
REFERENCES
- Lytzen R, Vejlstrup N, Bjerre J, Petersen OB, Leenskjold S, Dodd JK, et al. Live-born major congenital heart disease in Denmark: incidence, detection rate, and termination of pregnancy rate from 1996 to 2013. JAMA cardiology. 2018;3(9):829-37. [Crossref] [Google Scholar] [PubMed]
- Peyvandi S, Ingall E, Woyciechowski S, Garbarini J, Mitchell LE, Goldmuntz E. Risk of congenital heart disease in relatives of probands with conotruncal cardiac defects: an evaluation of 1,620 families. American journal of medical genetics Part A. 2014;164(6):1490-5. [Crossref] [Google Scholar] [PubMed]
- Beauchesne LM, Warnes CA, Connolly HM, Ammash NM, Tajik AJ, Danielson GK. Outcome of the unoperated adult who presents with congenitallycorrected transposition of the great arteries. Journal of the American College of Cardiology. 2002;40(2):285-90. [Crossref] [Google Scholar] [PubMed]
- Graham TP, Bernard YD, Mellen BG, Celermajer D, Baumgartner H, Cetta F, et al. Long-term outcome in congenitally corrected transposition of the great arteries: a multi-institutional study. Journal of the American College of Cardiology. 2000;36(1):255-61. [Crossref] [Google Scholar] [PubMed]
- Witham AC. Double outlet right ventricle: a partial transposition complex. American heart journal. 1957;53(6):928-39. [Crossref] [Google Scholar] [PubMed]
- Dewaswala N, Bolanos MD, Bhopalwala H, Reda H, Leventhal A. Severe Symptomatic Aortic Stenosis in an Octogenarian with Congenitally Corrected Transposition of the Great Arteries. CASE. 2024;8(3):133-7. [Crossref] [Google Scholar] [PubMed]
- Pino PG, Pergolini A, Zampi G, Calicchia A, Chialastri C, Orsini FM, et al. Congenitally corrected transposition of the great arteries with severely stenotic bicuspid aortic valve in an adult: a case report. Echocardiography. 2012;29(1):E13-5. [Crossref] [Google Scholar] [PubMed]
- Presbitero P, Somerville J, Rabajoli F, Stone S, Conte MR. Corrected transposition of the great arteries without associated defects in adult patients: clinical profile and follow up. Heart. 1995;74(1):57-9. [Crossref] [Google Scholar] [PubMed]
- Connelly MS, Liu PP, Williams WG, Webb GD, Robertson P, McLaughlin PR. Congenitally corrected transposition of the great arteries in the adult: functional status and complications. Journal of the American College of Cardiology. 1996;27(5):1238-43. [Crossref] [Google Scholar] [PubMed]
Article Processing Timeline
| 2-5 Days | Initial Quality & Plagiarism Check |
| 25-35 Days |
Peer Review Feedback |
| 45-60 Days | Total article processing time |
Ethics & Policies
Editorial & Management
Useful Links
Journal Highlights
Open Access Journals
Journal Flyer
